在涉及1,300至1,500原子规模的高精度DFT基准测试中,实现了卓越水平的精确度,并能够支持单卡处理高达15万个原子的数据量。
精确模拟分子在药物开发、蛋白质结构研究和核酸功能分析等领域至关重要。然而,长期以来,这些领域一直受到“尺度与精度”之间矛盾的影响:高精度量子化学方法通常只能涵盖数百个原子,而更可扩展的经典力场则难以描述复杂分子间的相互作用及非局部效应。尽管机器学习力场显著提升了精度,在真实生物大分子场景中仍面临三大挑战:小分子数据占主导地位、显式溶剂与大规模体系样本稀缺;长程相互作用建模困难;以及对于大规模系统的推理吞吐量不足,难以实现稳定的工程应用。
最近,IQuest Research(至知创新研究院)的UBio团队发布了一款名为UBio‑MolFM的基础分子模型框架。该框架旨在满足生物大分子体系高精度模拟的需求,并通过数据集、架构和训练策略的设计协同实现了突破性的进展。团队构建了迄今为止最大的高精度DFT数据集——Ubio-Mol26,包含超过1700万条记录,其中最大分子含有1200个原子的数据。这些数据重点涵盖了溶液环境中的蛋白质、DNA/RNA片段以及细胞膜等生物大分子体系。基于这个庞大的数据库,团队使用了最先进的E2FormerV2类Transformer架构,并结合显式的长程相互作用建模,在多阶段课程学习框架的辅助下实现了卓越的精度和吞吐量表现。
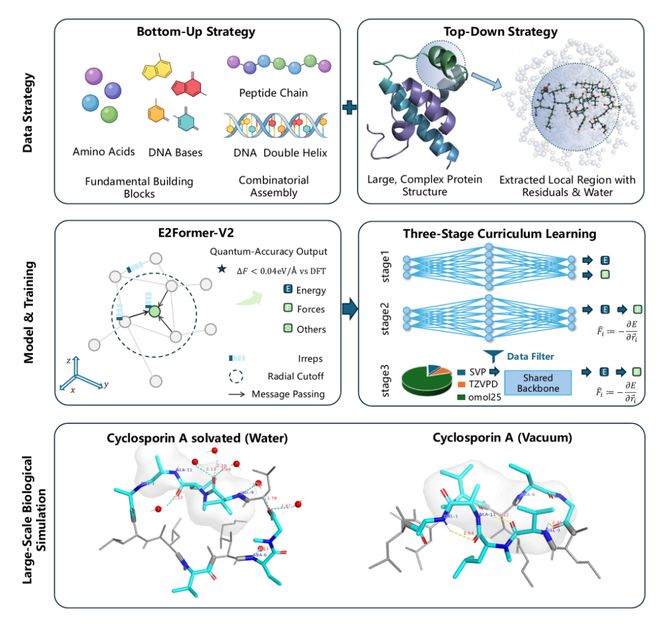
框架总览
01
此次发布的项目包括UBio-MolFM框架
Protein26数据集以及开源复现路径
UBio‑MolFM 是一套专为真实生物体系设计的基础分子模型框架,能够覆盖蛋白质、DNA/RNA、脂质膜及多种复合系统,并针对显式溶剂、长程耦合效应和大规模稳定推理进行了系统性优化。
UBio-MolFM 主要包括三个部分:一是用于生物系统的数据底座(UBio-Mol26/Protein26),二是大体系推理的等变架构实现(E2Former-V2),三是支持多理论层级融合训练范式,并提供开放代码、数据以及后续模型和工作流计划。
数据基础:更贴近生物系统
UBio-Mol26 包含大约1700万个构型,涵盖了蛋白质、DNA/RNA、脂质膜及复合体系的数据,且包括显式溶剂。每个单独的结构最大可达到1200个原子,并采用统一的过程生成高质量的DFT标注(wB97M-D3)。同时开放了一个标准化子集UBio-Protein26 5M(包含5百万训练和20万测试数据),其训练样本由4.5百万def2-SVP和50万def2-TZVPD组成;平均每个分子超过370个原子,以填补公共数据在生物大体系尺度上的空白。
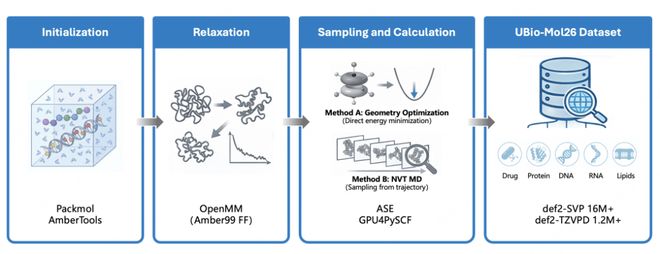
数据生成流程
数据生成方面,UBio-Mol26 采取了“自下而上枚举+ 自顶向下采样”的混合方法:通过系统地列举氨基酸短肽、核酸片段和脂质单元来确保基本组件的覆盖;同时从真实蛋白质结构中提取局部环境并进行溶剂化处理与化学封端,以增强对生物场景几何及相互作用模式的模拟。团队还提供了UBio-Mol26 与OMol25 的分布对比(t-SNE可视化),用于展示两套数据在特征空间中的互补关系。
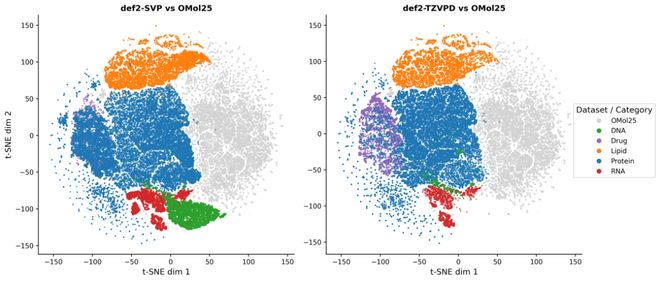
t-SNE 分析:UBio-Mol26 与 OMol25 对比
模型介绍:E2Former‑V2(线性扩展的等变Transformer)
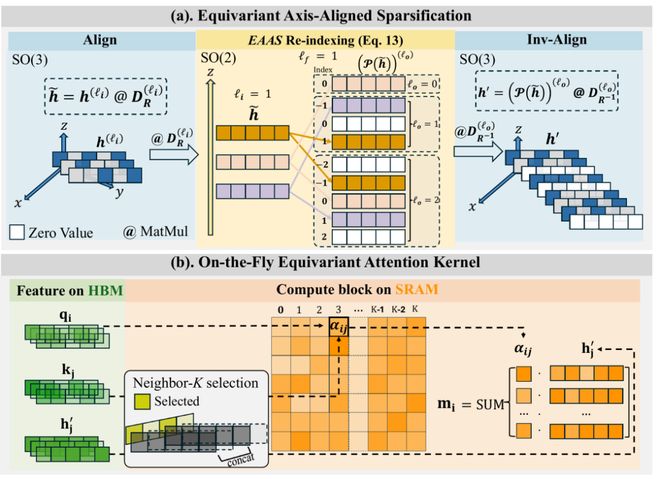
UBio-MolFM 使用了E2Former-V2架构,该架构通过“节点中心、硬件友好”的实现方式减少了稀疏边操作,提升了内存局部性,从而降低了大规模系统推理的成本;同时通过EAAS方法减少SO(3)张量积的开销,并结合LSR技术处理长程与短程相互作用。
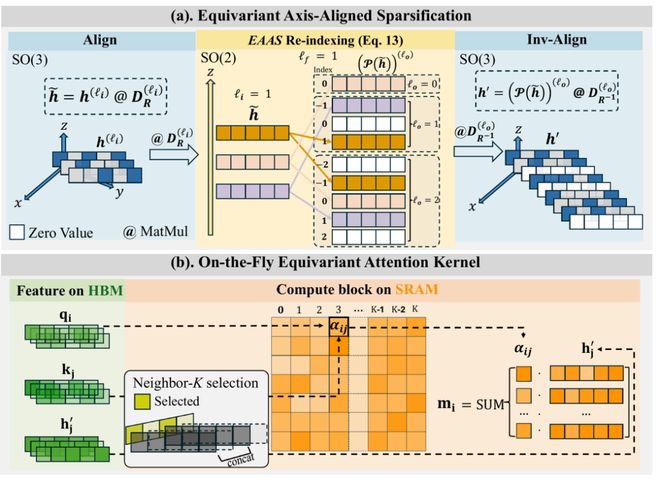
E2Former-V2 架构图解
训练过程:三阶段课程学习(多理论层级融合)
采用三个阶段训练,平衡了化学覆盖、物理一致性和尺度扩展:
第一阶段使用OMol25进行快速能量初始化,并通过并行预测能量和独立力头来提升吞吐量;
第二阶段舍弃了独立力头,力由能量梯度计算得出,以增强能量与力的一致性;
第三阶段引入UBio-Mol26的多保真数据集,并通过双头结构和仅用力损失进行监督处理不同理论层级的数据:SVP与TZVPD分别对应不同的能量头,而TZVPD只施加力损失以避免能量偏差。同时使用OMol25、SVP、TZVPD 8:1:1的比例以及相似性过滤保持训练的稳定性。
02
在生物大规模体系中实现了
“可引用精度”和“大规模吞吐”
推动高精度模拟更接近实际应用场景
对于科研及产业来说,分子基础模型需要在日常工作中发挥作用,必须回答以下两个问题:第一,在更接近真实的生物体系中,误差是否仍然可控且可验证;第二,在需要长时间轨迹和高频推理的分子动力学场景下,吞吐量能否满足工程使用需求。UBio-MolFM 的关键价值在于,它在同一套评估标准和技术限制下同时解决了这两方面的问题。
外推精度:在1,300至1,500原子规模上表现出显著优势
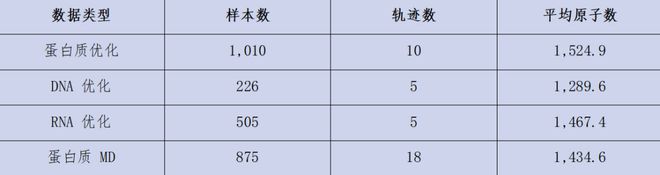
团队创建了一个外推测试集,涵盖蛋白质优化、DNA优化、RNA优化和蛋白质MD等多种任务,并使用MACE-OMol与UMA-S-1p1进行了对比。此测试集中样本的数量及平均原子数在文档中有详细统计:例如蛋白质优化包含1010个样本,每个样本的平均原子数量为1524.9;RNA优化则有505个样本,平均每个样品含有1467.4个原子。
外推测试集统计
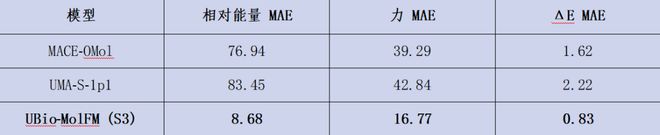
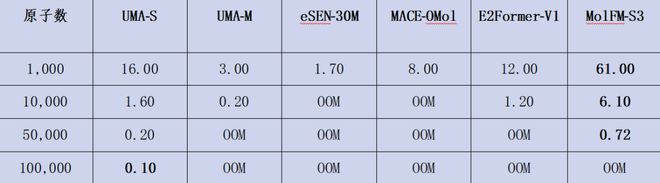
在代表性结果(蛋白质优化)中,UBio-MolFM 的表现显著优于其他模型:在相同条件下,MolFM-S3的吞吐量达到61 steps/s,相比之下UMA-S为16,MACE-OMol为8,E2Former-V1则为12;当扩展到十万原子规模时,MolFM-S3仍能保持6.10 steps/s的速度,而其他对比模型由于内存限制已经无法正常运行。这表明单卡可以实现最多处理15万个原子的推理。
在蛋白质优化任务中,UBio-MolFM在1,000至50,000个原子范围内展示了约4倍的吞吐量提升,将大规模计算从偶尔实验转变为可重复流程。这一成就使得“大体系计算”更加可行,并为进一步探索更有效的长程相互作用建模方式提供了明确方向。
结合外推精度、物理一致性和推理吞吐的表现来看,UBio-MolFM的定位更像是一个为生物体系高精度模拟提供的基础平台:一方面将可验证的生物系统模拟推向了更为真实的尺度(1,300至1,500个原子),另一方面通过Ubio-Mol26和E2Former-V2的设计协同工作,使模型能够同时处理复杂的生物结构、溶剂化环境及金属配位等关键相互作用。
你对UBio - MolFM有何看法?
生物体系里,很多「看上去像细节」的物理量,恰恰决定了模型能否用于解释与预测。UBio-MolFM 在文档中给出了多组物理一致性验证:
在纯水与 0.15 mol/L NaCl 溶液中,模型能够重现 RDF 结构与配位数,用溶剂统计量验证基本液体结构是否合理。
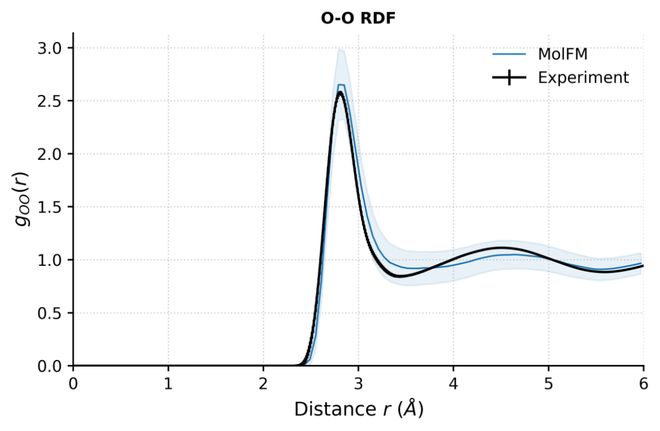
水的 RDF
在环孢素 A(CsA)体系中,模型能保持环境敏感构象:水中维持开放态、真空中维持闭合态,验证其对溶剂化环境变化的响应是否符合物理直觉。
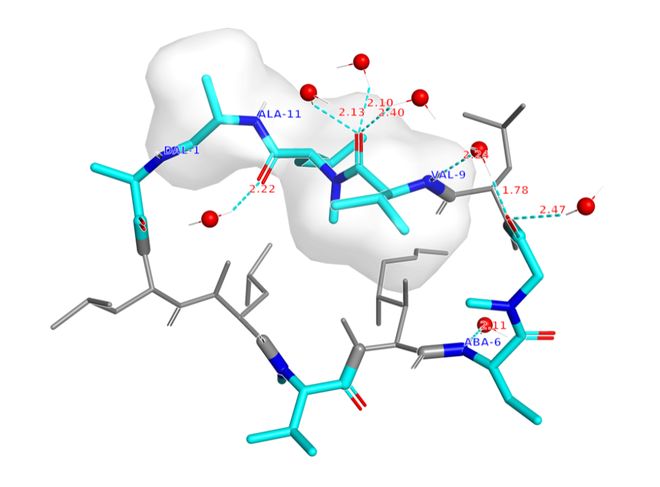
CsA 水中构象
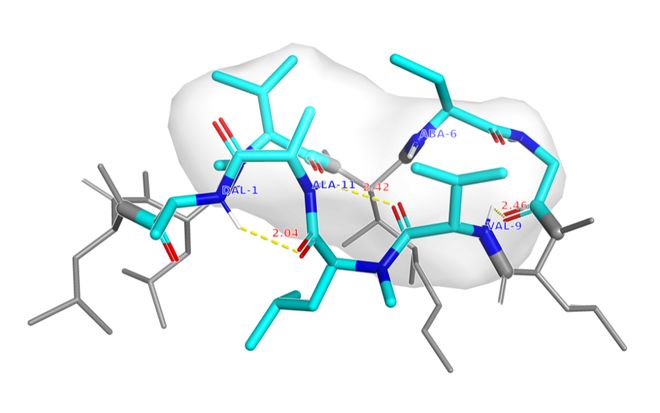
CsA 真空构象
在 RNA 1L2X + Mg²⁺ 系统中,模型重现 Mg–O 距离与角分布,体现对金属离子配位几何的刻画能力。对于核酸结构稳定性与功能相关研究,这类能力往往是「能不能用」的分水岭。
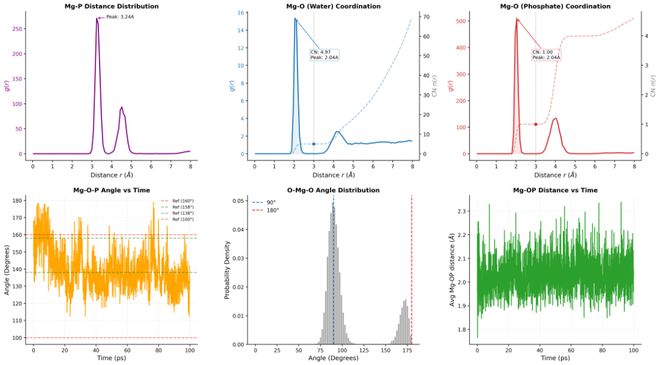
RNA Mg2+ 配位
推理吞吐:在1k–50k 原子范围内实现约4× 提升,把「大体系计算」从偶发实验推向可重复流程
在单卡 H100 上,UBio 团队对 MolFM-S3 与 UMA-S/UMA-M、MACE-OMol、eSEN、E2Former-V1 等等变模型做了推理吞吐测试(1k–100k 原子,保守力计算)。结果显示:在 1,000 原子规模下,MolFM-S3 为 61 steps/s,对比 UMA-S 的 16、MACE-OMol 的 8、E2Former-V1 的 12;在 10,000 原子规模下,MolFM-S3 仍有 6.10 steps/s,而多种对比模型已出现 OOM。单卡可实现至多 15 万原子的推理。
在 1k–50k 原子范围内,MolFM-S3 相对 UMA-S 约 4× 吞吐提升;在 100k 原子规模下,除 UMA-S 外大多数模型出现 OOM,UBio-MolFM 由于显式建模长程作用也未能幸免,这也提示了下一步的明确方向:探索更好的长程相互作用建模方式,进一步降低显存开销。
综合外推精度、物理一致性与吞吐表现,UBio-MolFM 的定位更接近「生物体系高精度模拟底座」:一方面把可验证的生物体系模拟推进到更真实的尺度(1,300–1,500 原子),另一方面通过 UBio-Mol26 与 E2Former-V2 的协同设计,使模型能够同时处理生物结构、溶剂化与金属配位等关键相互作用,从而为药物发现、蛋白构象动力学、核酸功能研究等提供统一的建模基础。
*头图
极客一问
你如何看待 UBio - MolFM ?